



Nell’immagine di copertina il biomatematico francese Jean-Claude Perez, già collaboratore del compoianto Luc Montagnier, e il cardiologo americano Peter McCullough
di Redazione Gospa News
Se il SARS-Cov-2 è stato palesemente creato in laboratorio sia nella sua variante più letale di Wuhan 1 che in quella Omicron più contagiosa ma anche più facilmente curabile senza necessità dei vaccini, il Vaiolo delle Scimmie (Monkeypox) è un virus di notevole complessità tanto da sembrare davvero un miscuglio demoniaco.
L’allarme lanciato dall’OMS per la sua nuova diffusione in Africa fa parte della solita enfatizzata propaganda allarmistica del direttore Tedros Adhanom Ghebreyesus, ex leader politico dei terroristi e golpisti etiopi del Fronte di Liberazione del Tigray, arruolato da Bill Gates come complice criminale di un progetto d’immunizzazione globale lanciato nel 1999 da Gavi Alliance con la Fondazione Rockefeller e promosso in seguito dal direttore di tale ONG Jens Stoltenberg, poi premiato come Segretario Generale NATO, in uno squallido intrigo tra Big Pharma e Lobby Armi.
Un Miscuglio Demoniaco dietro al Virus delle Scimmie
Appare infatti acclarato che la sua diffussione è ben più bassa di quella che creò l’allerta nel 2022 e che il contagio per via fisica e prevalentemente sanguigna, a differenza del virus della Covid-19 che è per via aerea, mette a vero rischio soprattutto gli omosessuali.
Ma la sua diffusione e recrudescenza (essendo apparso già a metà del XX secolo) trova ragione in una complessità di fattori: le mutazioni inspiegabili rilevati da due studi sulal sequenza genomica che lo rendono equiparabile a un agente patogeno potenziato in laboratorio, lo stretto legame con l’SV 40, una delle componenti cancerogene occulte del vaccino Pfizer, e la sua correlazione palese con la distruzione del sistema immunitario innescata proprio dai sieri genici mRNA che ha centuplicato in modo esponenziale i casi di risorgenza di di altri virus anche letali come l’Herpes Zoster.
Nel precedente articolo ci siamo concentrati sul ruolo svolto dall’SV 40, il Simian Virus, il virus di vacuolizzante della scimmia che provoca sovente infezioni latenti.
Le Tracce di Mutazioni da Laboratorio GOF
Oggi ci concentreremo su due studi sulle anomalie genomiche del Monkeypox
: quelli pubblicati nel 2022 dal nostro amico scienziato Jean Claude Perez, già braccio destro del compianto biologo Nobel Luc Montagnier ed autore di rivoluzionari studi sulle gravi reazioni avverse neurocerebrali dei vaccini Covid, e da Saathvik R. Kannan (Bond Life Sciences Center, University of Missouri, Columbia, US).
Il primo è stato riportato in auge da una brillante inchiesta sul tema di un Blog Francese. Il secondo è stato rilanciato dal noto cardiologo americano Peter McCullough nel suo Substack Courageous Discourse in cui ogni giorno analizza le manipolazioni delle campagne vaccinali da paerte di governi e Big Pharma.
«Un rapporto di Kannan et al ha indicato che ciò che è accaduto nel 2022 quasi certamente è avvenuto a causa di una mutazione con “Guadagno di Funzione” in uno o più geni all’interno del virus del vaiolo delle scimmie. Gli autori non speculano se ciò sia avvenuto in laboratorio o in natura» scrive McCullough menzionando la tecnica di potenziamento dei virus GOF (Gain of Function) utilizzata da Amthony Fauci sia nei test sul virus dell’aviaria che soprattutto nei pericolosi esperimenti sui coronavirus da cui è nato il SARS-Cov-2 e poi ripercorre la “storia americana” della contenuta epidemia.
La Diffusione del Virus soprattutto tra i Gay
«Il presidente Biden e il segretario dell’HHS Becerra hanno dichiarato un’emergenza sanitaria pubblica per il vaiolo delle scimmie negli Stati Uniti nel 2022 e hanno lasciato scadere la dichiarazione all’inizio del 2023. La crisi ha suscitato poca paura nell’americano medio poiché si è appreso che l’infezione era in gran parte un’eruzione pustolosa che si manifestava con intenso contatto sessuale tra uomini gay/bisessuali con vesciche cutanee attorno all’ano, sui glutei o sulla bocca» aggiunge il cardiologo blogger.
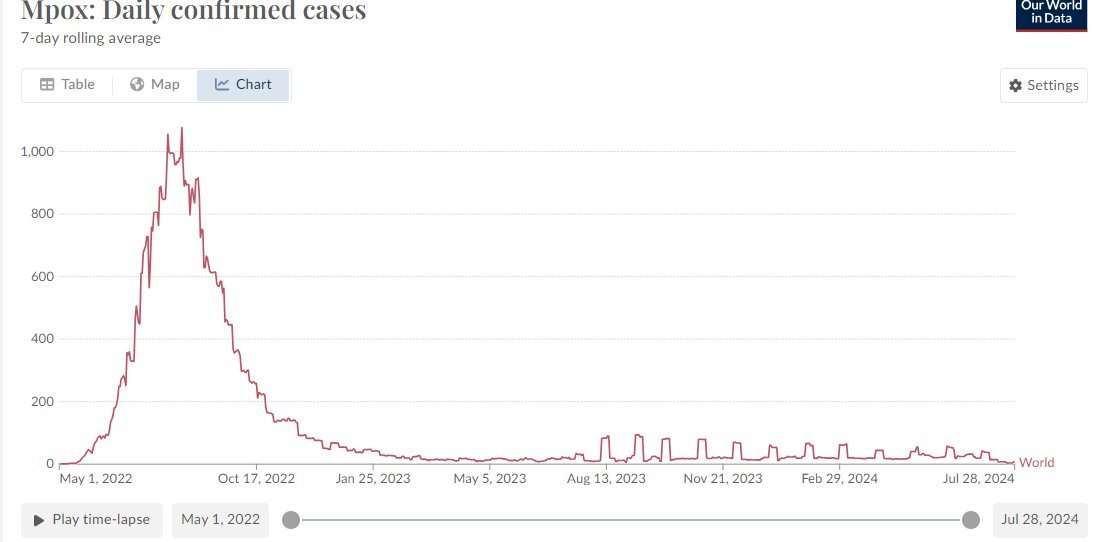

La stima è intorno all’84 % di diffusione proprio tra gli omosessuali.
«Il CDC ha controllato i test PCR, il farmaco primario tecovirimat e il vaccino Bavarian Nordic Jynneos. A partire dal 10 gennaio 2024, il CDC ha interrotto l’aggiornamento dei dati sul vaiolo delle scimmie a causa del basso livello di trasmissione del sottotipo clade 2 negli Stati Uniti che ha provocato 32.063 casi e 58 decessi, in gran parte tra gli uomini con malattia da HIV avanzata» rammenta McCullough.
Alle stesse conclusioni è giunto il sito Le Blog de Patrice Gibertie in cui si rammenta che «questa malattia è apparsa negli anni ’50 nelle scimmie e il primo caso nell’uomo risale al 1970. Di solito il primo sintomo a comparire è la febbre, seguita da “un’eruzione cutanea, che spesso inizia sul viso e può diffondersi ad altre parti del corpo, compreso il palmi delle mani, piante dei piedi e genitali”».
Il blogger cita molteplici fonti spiegando che «a livello globale, il tasso di sopravvivenza dopo aver contratto l’infezione da virus del vaiolo delle scimmie è del 99,942%. Degli 80.850 casi di vaiolo delle scimmie registrati dal 2022, solo 55 sono morti».
Le Manipolazioni Genomiche scoperte dal professor Perez
Infine arriva al nocciolo della questione: «Il vaiolo delle scimmie NON è un virus presente nell’aria e non lo sarà mai – a meno che una mente criminale non lo aiuti! E richiederà competenze geniali nella biologia molecolare».
Purtroppo le tracce di questo “principio di manipolazione” sono state scoperte proprio da Perez che aveva denunciato insieme a Montagnier l‘origine artificiale del SARS-Cov-2 contenente sequenze di aminoacidi dell’HIV.
Gli autori dello studio sono Jean-Claude Perez e Valère Lounnas del Laboratorio Europeo di Biologia Molecolare. Il loro studio è intitolato “May 2022 : Peculiar Evolution of the Monkeypox Virus Genomes – Maggio 2022: particolare evoluzione dei genomi del virus del vaiolo delle scimmie”.
«Gli scienziati hanno confrontato l’evoluzione di 14 genomi del virus del vaiolo delle scimmie con l’obiettivo di scoprire mutazioni o altre evoluzioni virali (ricombinazione) che potrebbero spiegare l’impatto improvviso di questa epidemia circolante a bassissimo livello» scrive Patrice Gibertie.
Per caso gli scienziati hanno scoperto che il cosiddetto virus circolante del vaiolo delle scimmie contiene una sequenza lunga 30 T al centro del genoma del vaiolo delle scimmie, tra l’RNA dipendente dal DNA e la proteina di inclusione del vaiolo bovino di tipo A.
Perché è strano? Si chiede il blogger francese che poi risponde:
«Ebbene, secondo gli scienziati, è perché questo gene non è mai completamente presente in una sequenza. Gli scienziati affermano che mentre questi risultati possono essere comuni alla fine di un genoma, ad esempio alla fine del virus dell’encefalite delle scimmie, non sono quasi mai presenti completamente in una sequenza».
«Per la prima volta nella storia, si ritiene che il virus del vaiolo delle scimmie circoli contemporaneamente in diversi paesi del primo mondo. Questo virus avrebbe subito più di 50 mutazioni nell’arco di 4 anni invece dei 50 anni che avrebbero dovuto impiegare. E ora sappiamo che contiene qualcosa che non dovrebbe esserci» conclude il blogger francese.
Insomma, viste le conclusioni di Perez e McCullough, si tratta con ogni probabilità dell’ennesimo caso di un virus potenziato in laboratorio per poter rilanciare un allarme enfatizzato in modo terroristico per continuare il business infinito delle Big Pharma su vaccini anche in questo caso pericolosi per le molteplici e gravi reazioni avverse…
Vista la complessità dei due studi sulle anomale mutazioni dei virus ci limitiamo a riportare gli Abstract senza commenti specifici ma con relativi link per la lettura delle ricerche complete.
Redazione Gospa News
© COPYRIGHT GOSPA NEWS
divieto di riproduzione senza autorizzazione
segui Fabio Carisio su Twitter
e Gospa News su Telegram
IL SOMMARIO DELLO STUDIO DI PEREZ
Confrontiamo l’evoluzione di 14 genomi del virus del vaiolo delle scimmie fino a quello del maggio 2022 che si sta attualmente diffondendo tra gli esseri umani in numerosi paesi al di fuori dell’Africa. Il nostro scopo era quello di scoprire mutazioni o altre evoluzioni virali (ricombinazione) che possano spiegare l’impatto improvviso di questa epidemia circolante a bassissimo livello o allertare su un potenziale carattere patogeno peculiare. Abbiamo evidenziato la presenza di un gran numero di basi T in successione, a livello della polimerasi, tra la subunità rpo132 della RNA polimerasi DNA-dipendente e la proteina di inclusione di tipo A del vaiolo bovino, progressivamente ascendenti dall’assenza di un pattern caratteristicamente lungo di basi T in successione (≤ 10) nei primi genomi del 1971, fino a 19 basi T nel ceppo di riferimento Israel 2018 e successivamente 30 basi T nei ceppi 2022.

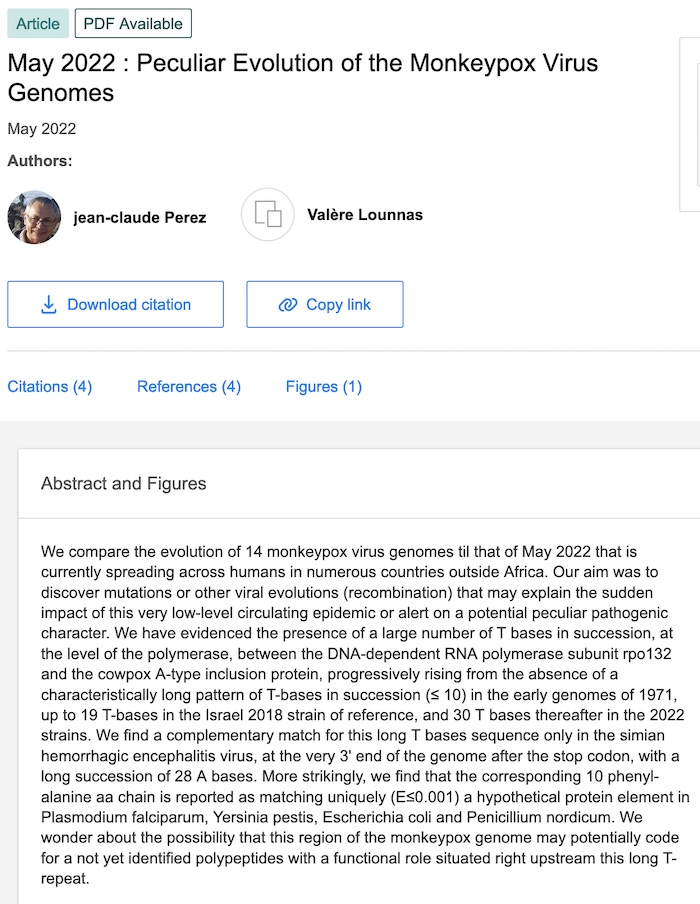
Troviamo una corrispondenza complementare per questa lunga sequenza di basi T solo nel virus dell’encefalite emorragica scimmiesca, all’estremità 3′ del genoma dopo il codone di stop, con una lunga successione di 28 basi A. Ancora più sorprendente, troviamo che la corrispondente catena di 10 fenil-alanina aa è segnalata come corrispondente in modo univoco (E≤0,001) a un ipotetico elemento proteico in Plasmodium falciparum, Yersinia pestis, Escherichia coli e Penicillium nordicum. Ci interroghiamo sulla possibilità che questa regione del genoma del vaiolo delle scimmie possa potenzialmente codificare per un polipeptide non ancora identificato con un ruolo funzionale situato proprio a monte di questa lunga T-ripetizione.
IL SOMMARIO DELLO STUDIO DI KANNAN
Delle 10 mutazioni totali nell’MPXV RC, 6 sono ritornate all’isolato del 1965. I428 si trova all’interno dell’inserto 2, che porta anche mutazioni di resistenza al PAA ma la mutazione di resistenza non è nella posizione 428. È possibile che I428T si sia evoluto come mutazione secondaria per compensare funzionalmente l’idoneità dei virus resistenti al PAA (C356T, G372D e G380S). Le mutazioni revertanti nelle posizioni 484 e 501 sono in stretta prossimità di L108. Pertanto, sembra che le mutazioni 484 e 501 siano ritornate per compensare la mutazione L108F.

Oltre alle mutazioni nel DNA RC discusse qui, anche altre proteine virali degli isolati 2022 contengono una varietà di mutazioni. Ad esempio, ci sono quattro mutazioni (V6D, D282 N, T601 M, D633E) nel gene della RNA polimerasi DNA dipendente (OPG151) negli isolati 2022. Tuttavia, a causa della mutazione nel DNA RC, non abbiamo discusso le mutazioni in altri geni di MPXV. È stata segnalata una struttura a bassa risoluzione di VACV composta da E9, A29, D4 e D5 [22]. È stato riportato anche un ortologo PCNA in VACV (G8R) [23]. La struttura a bassa risoluzione riportata non contiene G8R. È noto che PCNA e i suoi omologhi o ortologhi sono parte integrante del meccanismo di replicazione di molte specie. È molto probabile che anche VACV G8R e MPXV G9R facciano parte dell’RC in due poxvirus. La nostra modellazione strutturale mostra che G9R può essere facilmente sistemato tra F8L ed E4R. Inoltre, AlphaFold ha previsto accuratamente le eliche N-terminali di A22R, come visto nella struttura cristallina di VACV D4 in complesso con l’N-terminale A20 [25]. Queste eliche sono state utilizzate per la sovrapposizione per ottenere la posizione di E4R senza alcuna modifica nell’orientamento delle due eliche. È possibile che queste due eliche adottino un orientamento diverso, che potrebbe riposizionare E4R. Tuttavia, indipendentemente dalla posizione dell’E4R, rimane spazio sufficiente nell’RC per ospitare il G9R all’interno dell’MPXV G9R.
In sintesi, abbiamo presentato i componenti minimi del complesso di replicazione MPXV e identificato le mutazioni in questo complesso che probabilmente contribuiscono all’epidemia di MPX del 2022. La mutazione L108F in F8L, comparsa nell’epidemia del 2022, è vicina al nucleotide modello “capovolto” e migliorerà la processività di F8L, mentre W411L probabilmente migliora il legame di un componente del complesso di replicazione alterando la regione esposta superficiale. A causa dell’elevata sequenza e della somiglianza strutturale tra i poli virali del DNA di tipo B, si prevede che i trattamenti antivirali attualmente approvati mantengano l’efficacia contro l’attuale iterazione di MPXV. Tuttavia, l’evoluzione delle mutazioni di resistenza rimane possibile poiché i percorsi funzionali critici sono già stati suscettibili a mutazioni funzionali all’interno delle proteine virali.
FONTI PRINCIPALI LINK
